SYNTH-cancer: A Foundational Atlas
SYNTH-cancer: A Foundational Atlas
SYNTH-cancer
We created a foundational functional genomics cancer dataset with AI.
Cancer remains one of the most formidable challenges in medicine, characterized by its complex heterogeneity across tissue types, genetic backgrounds, and tumor microenvironments. The Cancer Genome Atlas (TCGA) has cataloged the molecular and genomic features of thousands of real tumor biopsy samples, providing invaluable insights into cancer biology. Much of modern molecular cancer biology relies on the data provided by TCGA.
We set out to see if we could recreate a foundational functional genomics dataset with AI that could augment or even supplant the huge effort that went into TCGA: an atlas of AI-simulated gene expression data of cancer and normal samples, spanning the same wide range of cancer types and anatomical sites. The model generating this AI data atlas was not trained on TCGA data, so TCGA data represents an appropriately independent point of comparison for the AI data.
This resource is designed to complement and extend existing tumor expression profile datasets by offering a resource with key advantages:
- A workflow that creates sample data without the batch effects from performing sequencing in different centers at different times
- Samples which are ethically unobstructed since they are not related to specific human subjects
- Complete control over the demographics, tissues, cancer types, and numbers of samples generated (although here we mirror TCGA to facilitate direct comparison)
- The normal samples do not represent biopsies of somewhat normal tissue adjacent to a tumor, but rather entirely normal tissue.
Building the AI Atlas
We used the synthesize.bio v2.2 model to make a complete expression dataset, in a single technical batch, for samples matching those of TCGA in tissue, disease type, sex, age, race, and ethnicity.
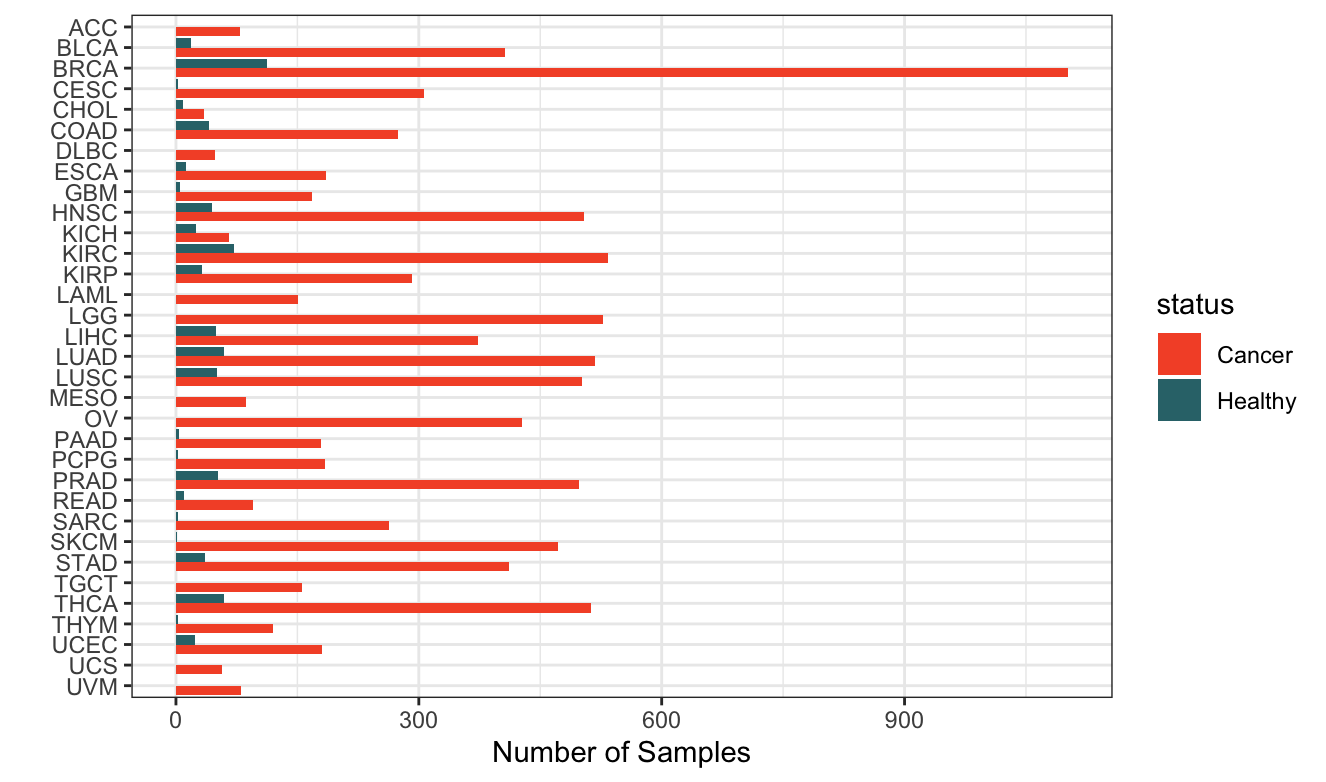
Figure 1. Data for almost ten thousand samples! A total of 10523 samples are synthesized and analyzed, both cancer and healthy normal, across all TCGA cancer types.
Hierarchical clustering across the full AI-generated dataset shows a high degree of internal data structure and clustering of genes specific to given organs.
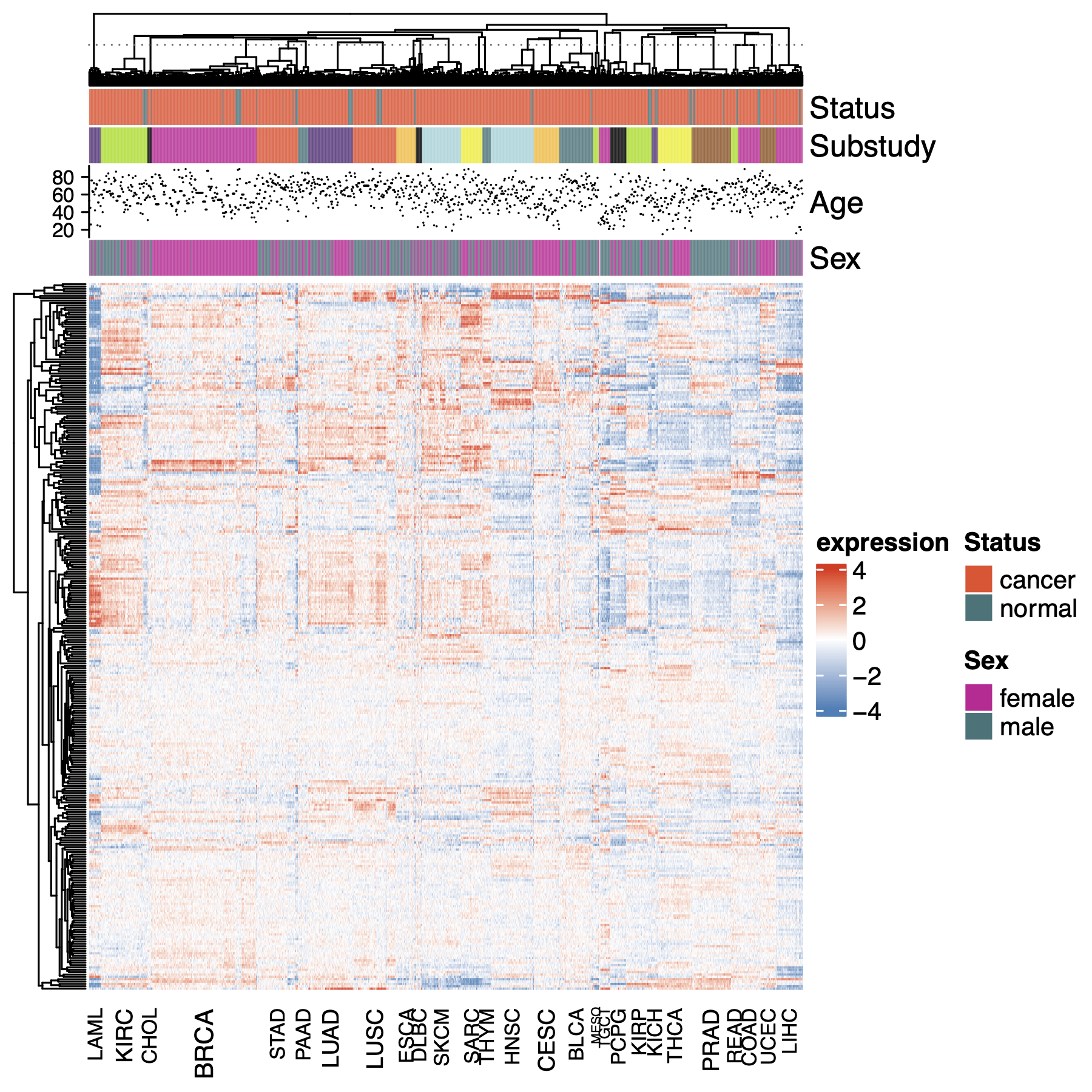
Fig 2. The AI data shows great structure. A heatmap of AI expression data surveying the whole dataset. Samples are clustered first within each substudy, then the means of the substudies are clustered. For ease of computation, samples are a random selection of 25% of all samples in the dataset. Genes are the top 1000 most variable across all tissues. Clustering in both dimensions is agglomerative on scaled euclidean distance with mean summarization.
The Head-to-Head Test: AI vs. The Lab
How does AI-generated expression data cluster with respect to lab data? Tissues/organs should be the strongest factor, and ideally cancer status is a stronger factor in clustering than whether the data is from AI or a lab. To assess this we have performed UMAP analysis of up to 100 samples in each organ/cancer/source group and plotted the same coordinate data twice, colored first by cancer status and then again colored by data source (AI vs. lab).
We see here that tissue of origin is indeed the strongest factor, and reassuringly, LUAD (lung adenocarcinoma) and LUSC (lung squamous cell carcinoma) are almost congruent. Cancer status and data source can be compared by focusing on the point color of these two plots
Some organs, like kidney and lung, show quite strong separation between cancer and adjacent normal samples. Others, like prostate, show clear separation by data source as well as by cancer status. This could reflect factors like disease stage or metastasis status in AI vs. lab data since in this dataset these variables are not specified in the AI data generation… an interesting avenue to explore.
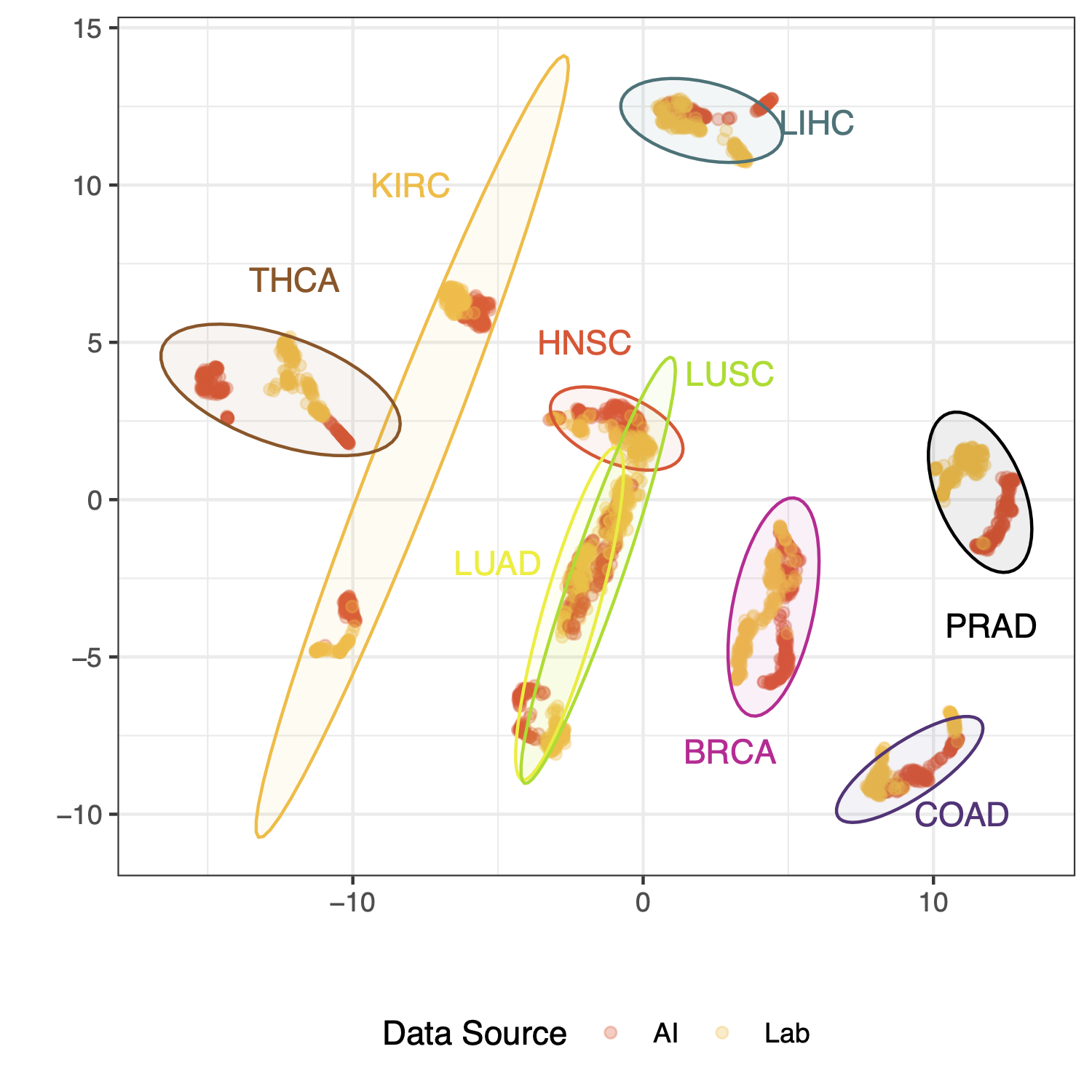
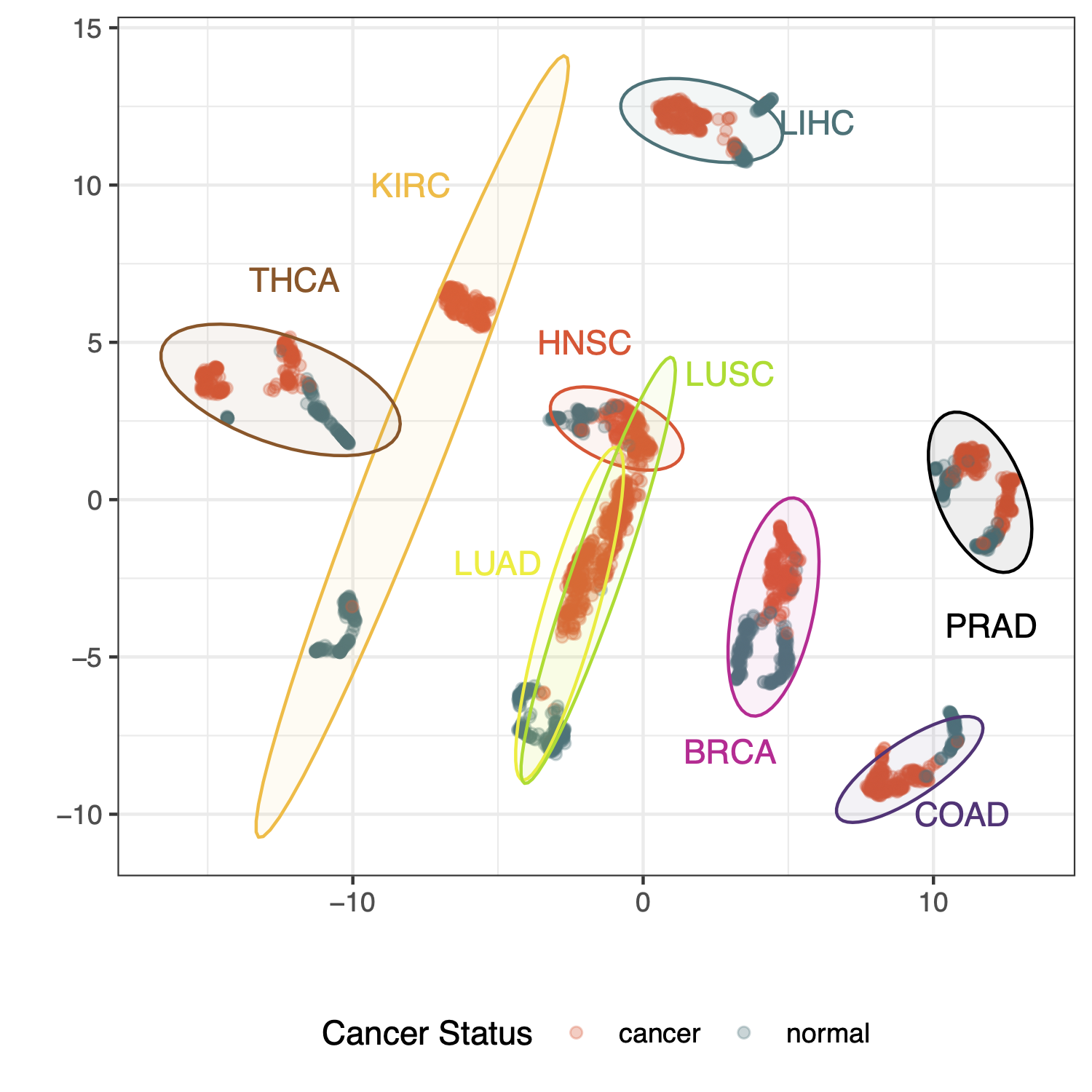
Fig 3. AI and lab data look really similar, and the biology of the data shines through. UMAP clustering results of expression data for up to 100 samples per group; where a group is a unique set of tissue of origin, cancer disease status, and data source (lab or AI). Gene data used for clustering are the top 1000 most variable across all tissues of origin and cancer status.
AI Data Can Do Molecular Subtyping
The heterogeneity of cancers within many traditional pathological cancer types has been recognized to cluster into a few distinct subtypes based on molecular features like gene expression and somatic genetic variation, and these subtypes are often associated with important clinical prognoses, etiologies, and differential responses to specific cancer treatments. Two subtyping schemes that were pioneers in this approach and have yielded important insights into their respective cancer areas are the PAM50 subtypes for breast adenocarcinoma and the consensus molecular subtypes (CMS) for colorectal carcinoma. Here we apply these subtyping schemes to AI and lab data to compare their distributions and balance.
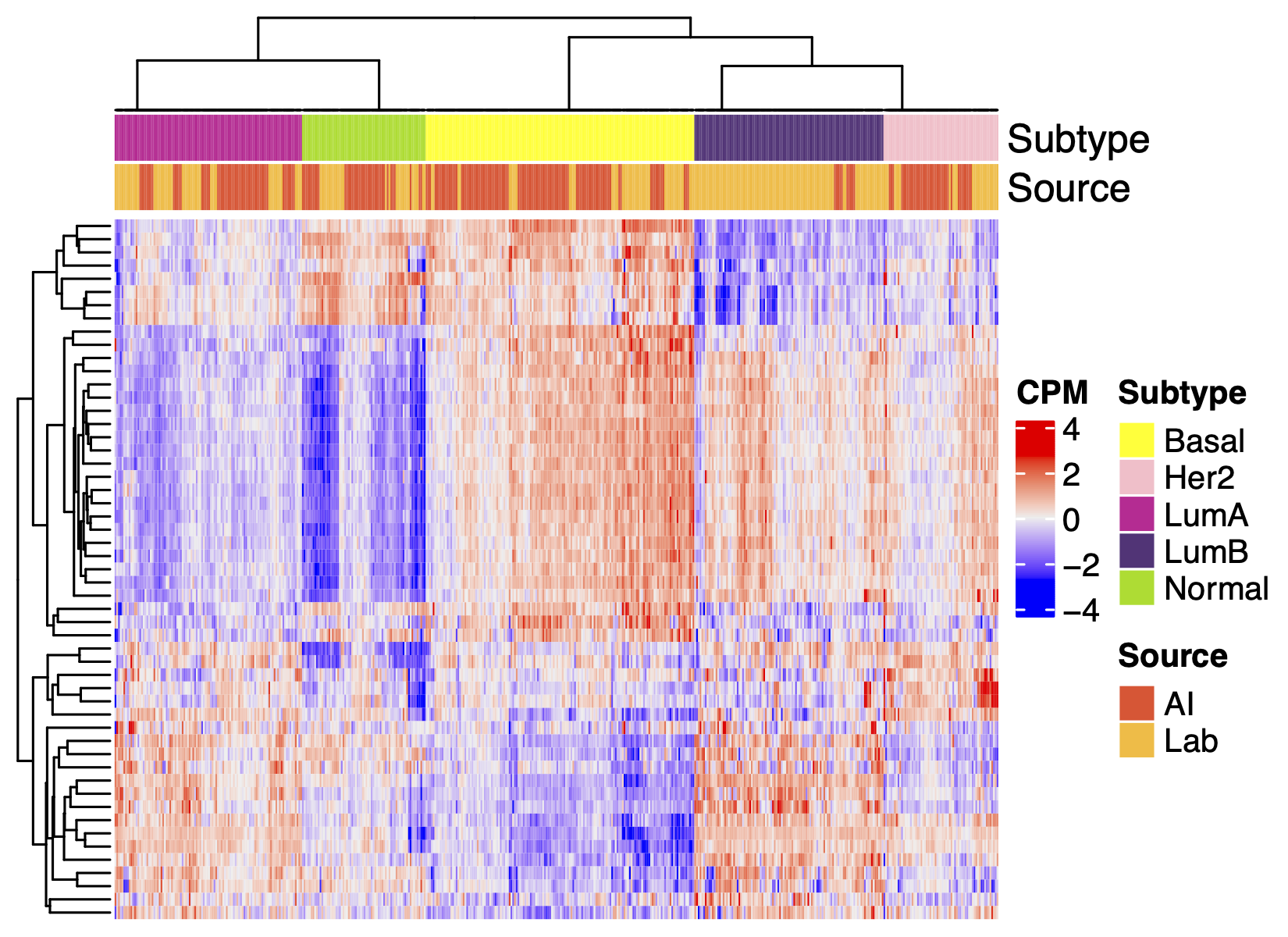
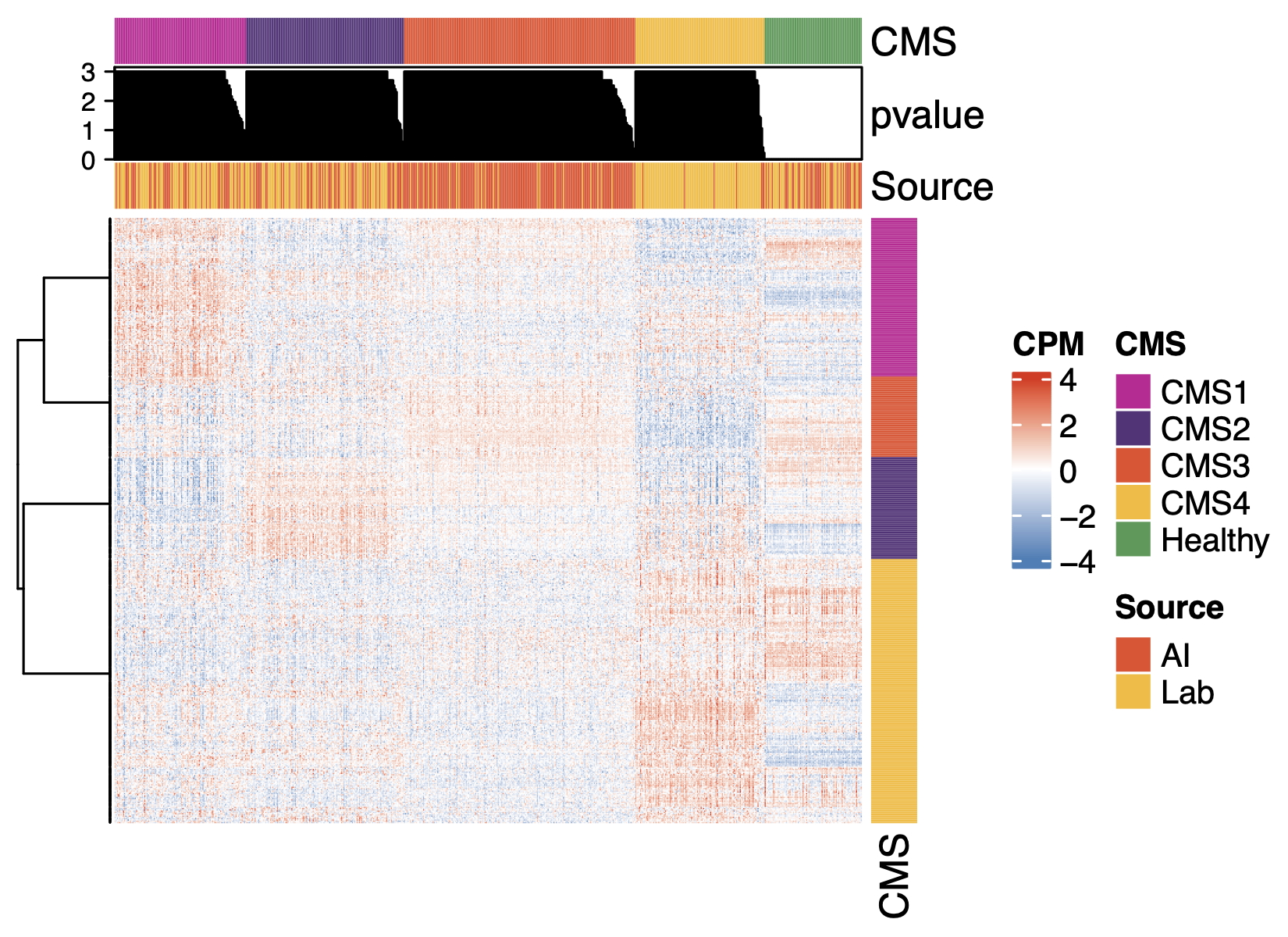
Fig 4. Molecular subtyping of AI data works well. Clustering of genes that define the molecular subtypes of breast cancer (PAM50) or colon cancer (CMS). Subtype assignment is performed according to canonical published methods, hence the differences in the features of the figures (p-value, gene annotation, etc.).
Despite some difference in the incidence of the subtypes in the two data sources (e.g. CMS4 being more abundant in AI data), these heatmaps show that AI-generated gene expression data generally has essentially the same molecular subtyping features as lab-generated data: the correlation structure of the biomarkers of the subtypes and the distributions of the subtype signatures across samples.
Does AI See the Bigger Biological Picture?
As a core analytical method, it’s key that differential expression of AI data be fairly comparable to lab data. To assess this, we contrast lung adenocarcinoma samples with adjacent normal lung samples using DESeq2 and find a strong similarity between AI and lab results on an individual gene basis.
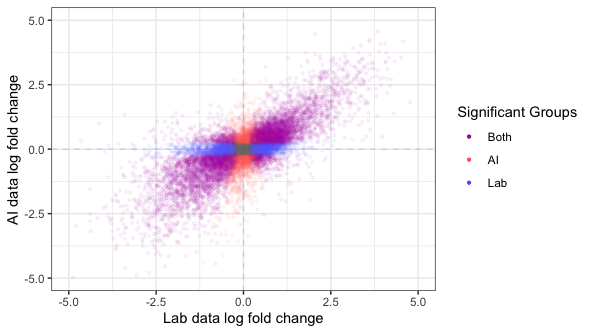
Fig 5. Differentially expression in AI data looks much like in lab data. DESeq2 differential expression results contrasting lung adenocarcinoma samples with lung adjacent normal samples. Points are genes, colored by whether the result for each gene is statistically significant in one or both groups at a nominal p-value of 1e-6.
Analysis of trends in gene sets can be a valuable extension to gene-level differential expression, and should also be comparable between AI and lab data. Performing testing for functional enrichment of gene sets in each of these differential expression results, we see reassuringly similar results.
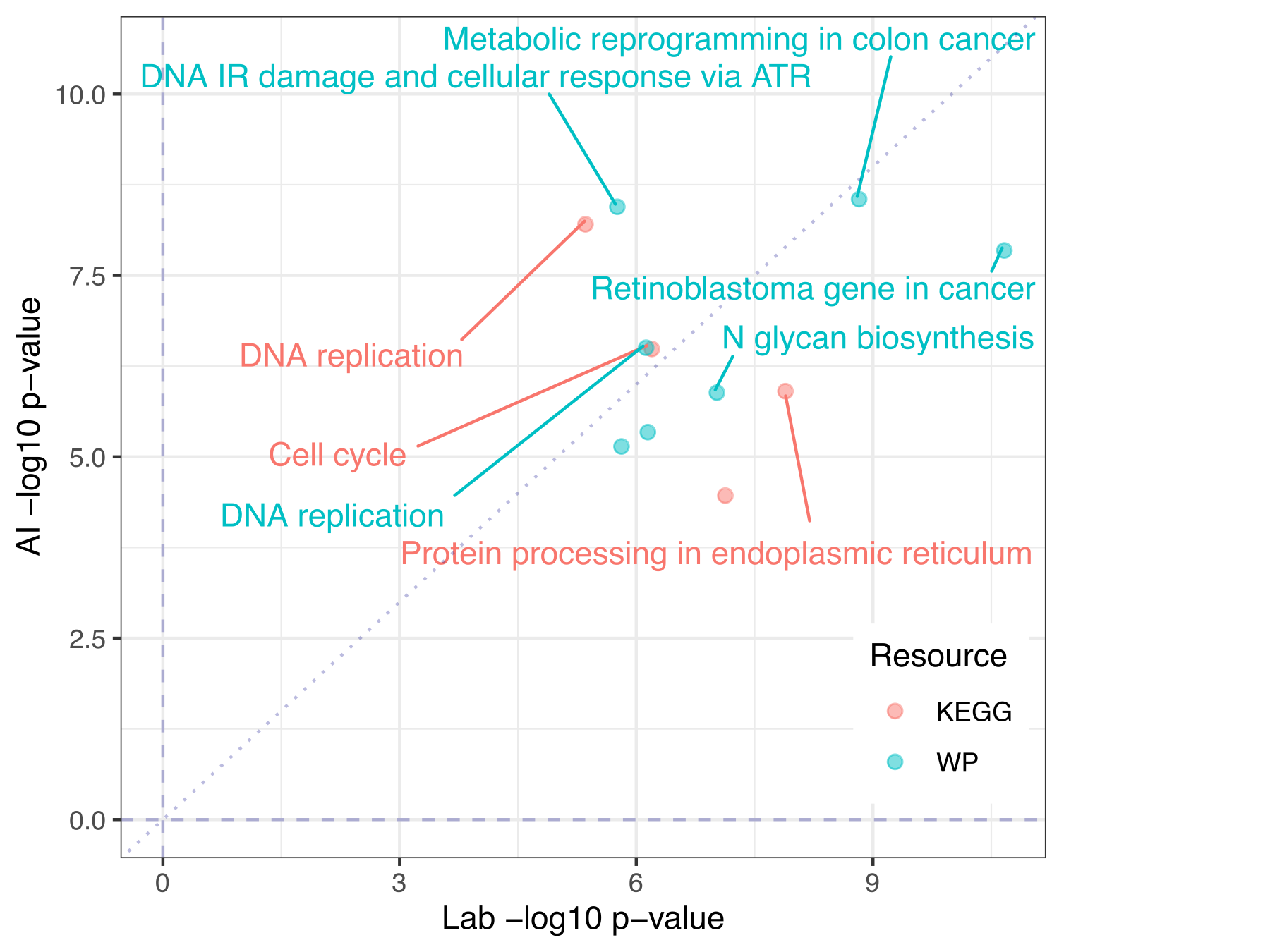
Fig 6. Gene set results in AI data look much like in lab data. KEGG and WP gene set top hits from enrichment analysis of lab or AI differential expression of lung adenocarcinoma vs. normal adjacent lung. Gene sets selected to be shown and/or labeled by arbitrary combined p-value threshold.
Putting Key Cancer Genes Under the Microscope
Expression patterns of certain individual genes are key to the understanding and precision treatment of cancer. They can be functional readouts of the driver mutations of cancer, biomarkers predicting treatment response or innate/acquired resistance, or simply markers useful for detection and prognostic evaluation. It’s clear that AI-generated data must reliably deliver fairly accurate readouts of relevant genes to be a useful resource.
Here are a few hand-picked genes for which we hoped that AI-generated data had features closely resembling lab data, and the upshot is that they largely succeed.
It’s important to note that there were no instances where we selected a gene, examined the data, and elected not to highlight it here because the results were disappointing.
ERBB2
The ERBB2 gene encodes the Her2 protein, a protein highly expressed on the surface of a subset of breast cancers (“HER2 positive”) and the target of Herceptin and other therapeutic antibodies.
The elevated expression in some breast cancers is seen in AI-generated data, as is the tumor upmodulation in various other tissues that have supported pursuit of indication expansion for anti-Her2 therapeutic agents.
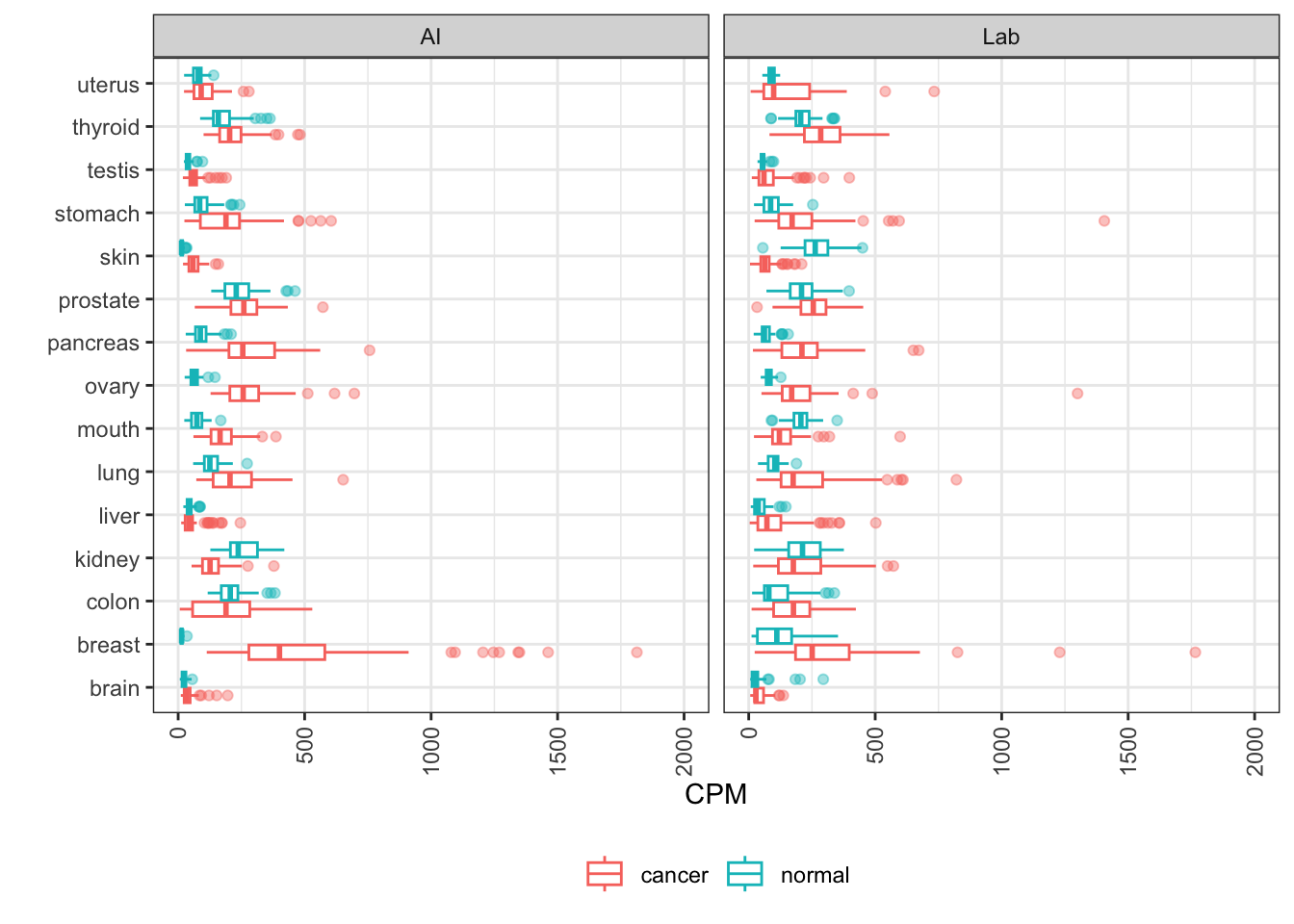
MUC1
MUC1 encodes the mucin 1 transmembrane glycoprotein, which plays a role in metastasis and treatment resistance in multiple cancers.
Key elements here are the relatively high expression in both gastric tumors and healthy stomach, and elevated expression in a portion of pancreatic, ovarian, lung, and breast cancers. These features are well conserved in AI-generated data.
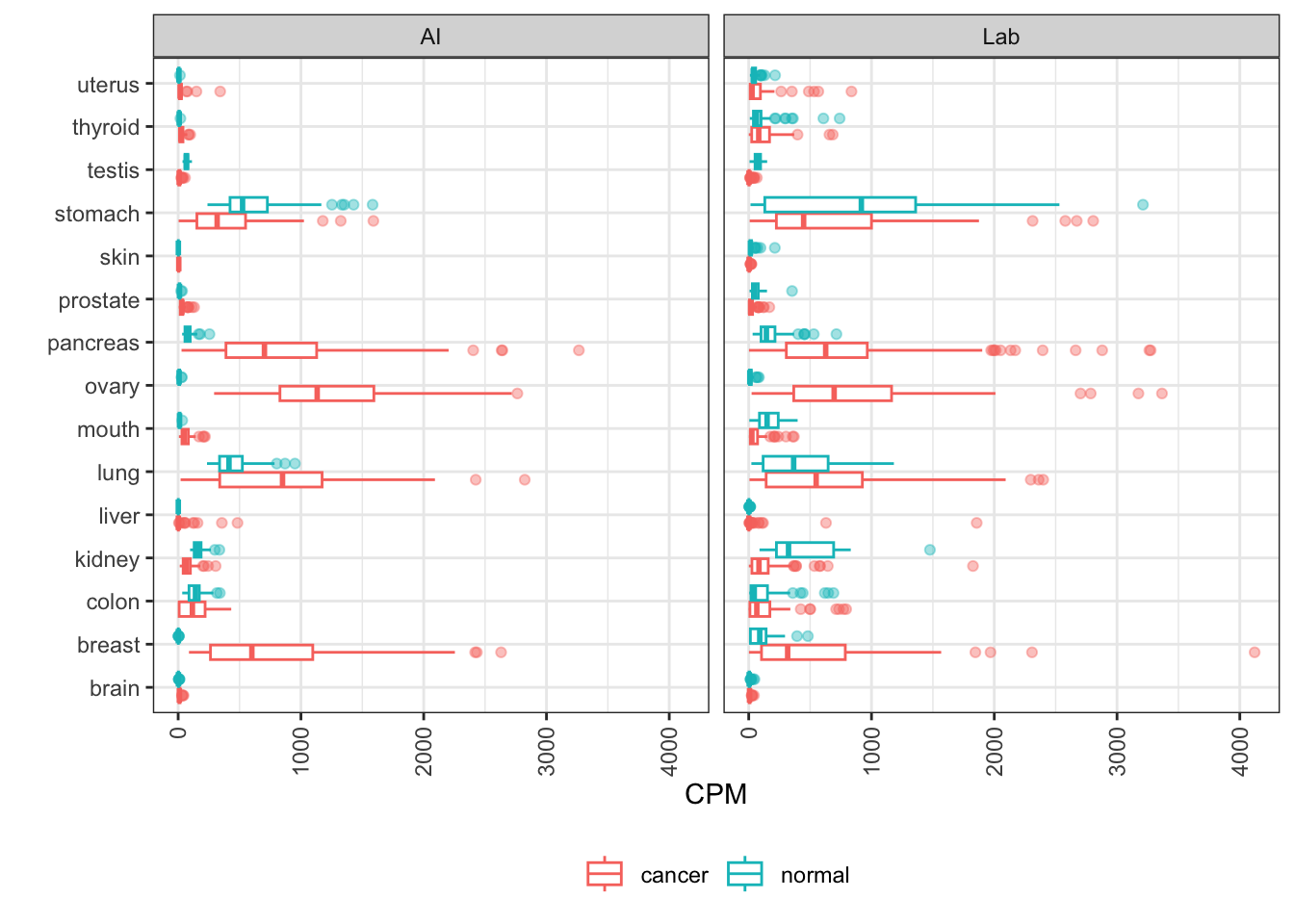
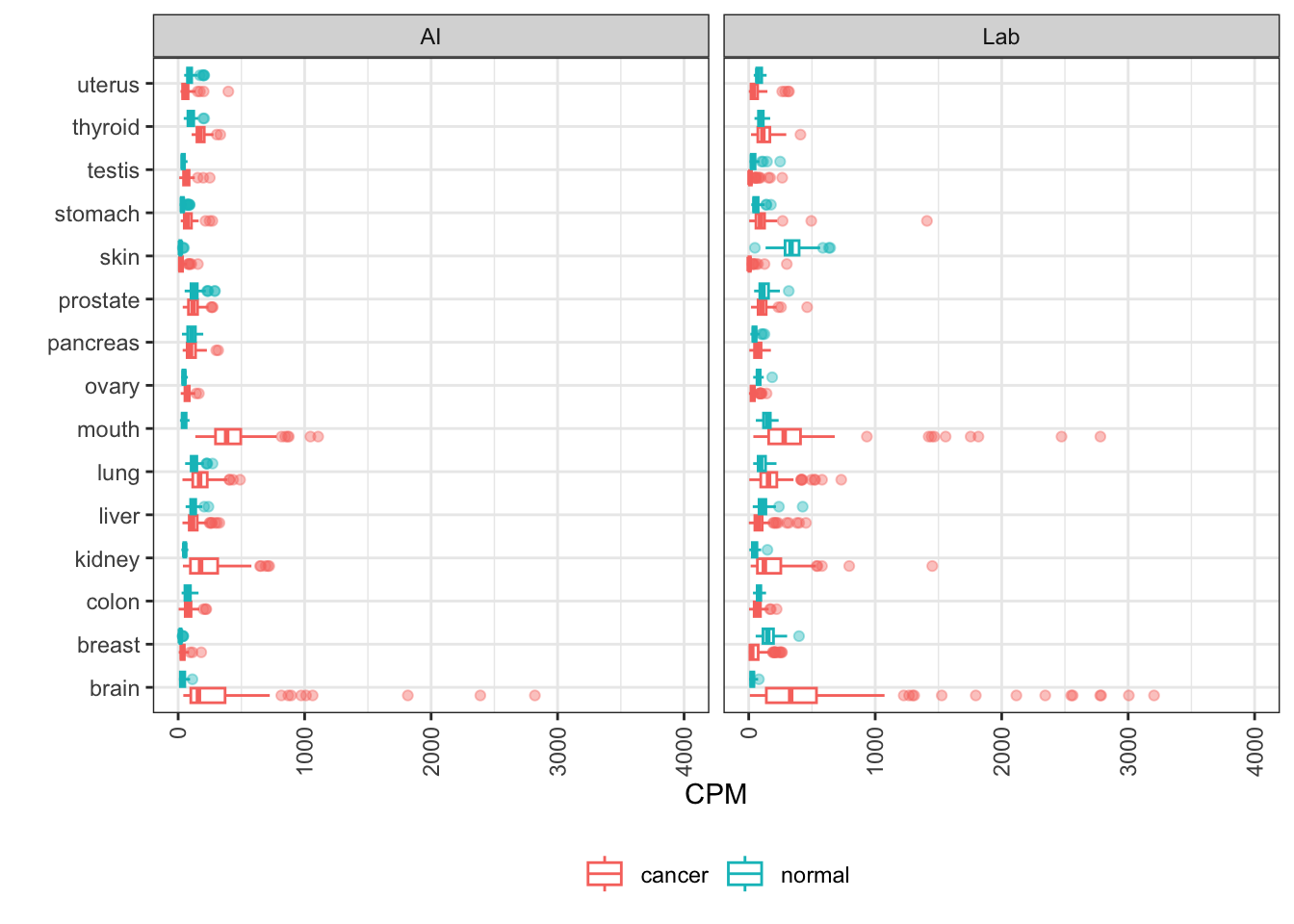
MLANA
MLANA encodes melan-A, also known as MART-1 (melanoma antigen recognized by T cells 1). A cell surface protein, it’s being investigated as a target for melanoma cancer vaccines.
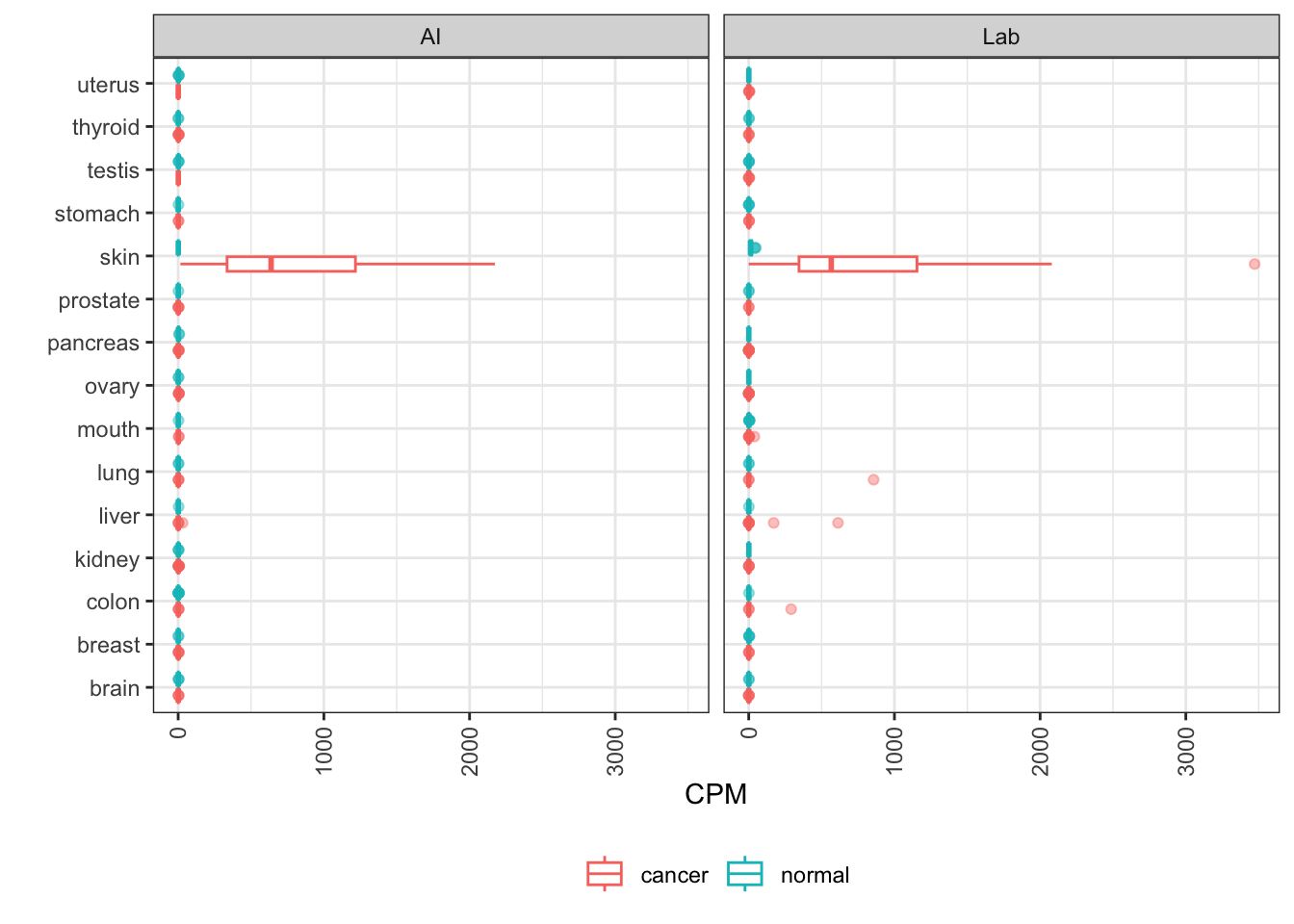
CEACAM5
CEACAM5 encodes CEA, a blood glycoprotein secreted from colorectal carcinomas and other tumors. It serves as an analyte for cancer screening tests.
The elevation in colon cancer and the overlap of that distribution with normal colon expression is fairly consistent between AI-generated and lab data, albeit a little closer in AI data.
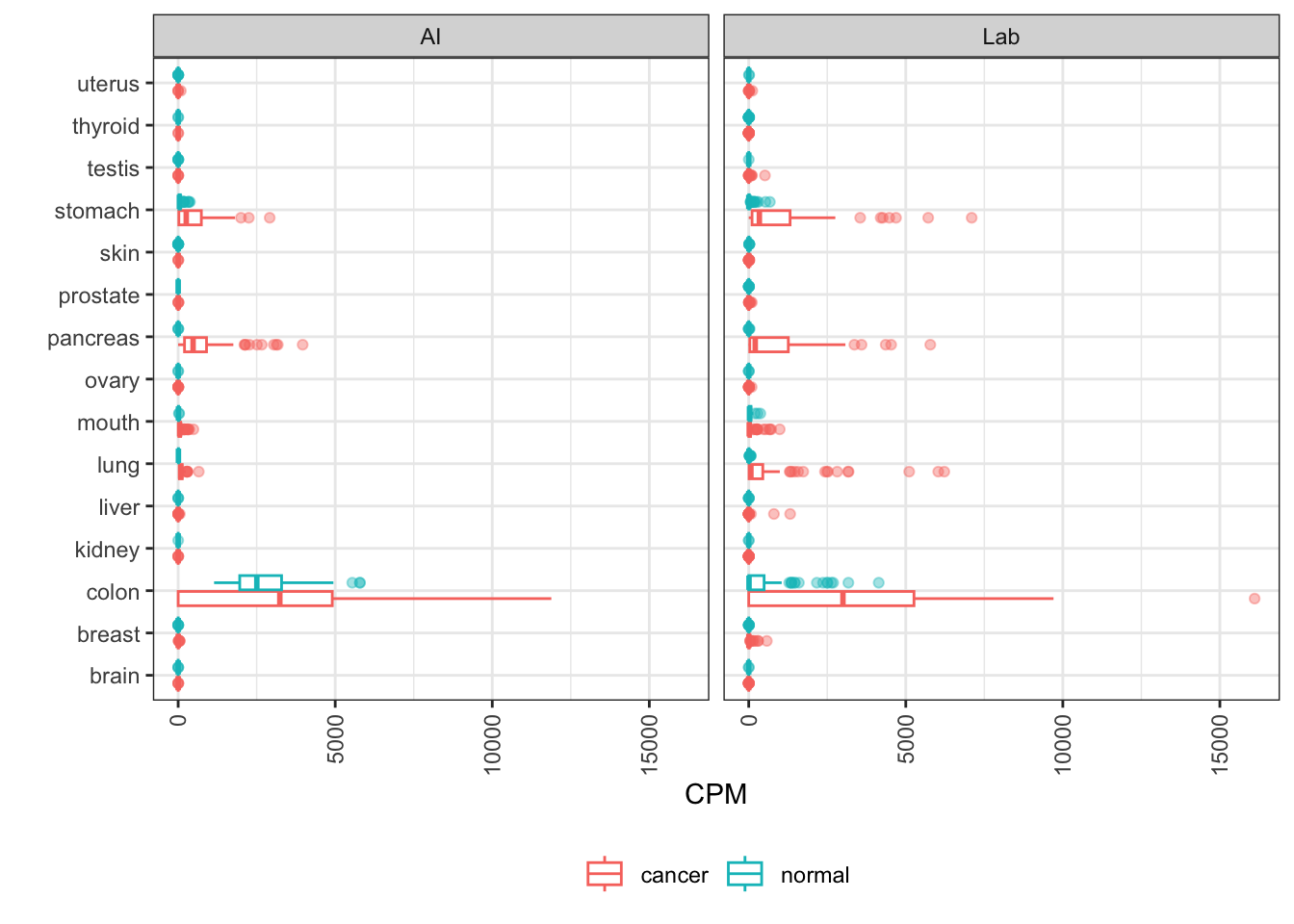
EGFR
EGFR (Epidermal Growth Factor Receptor) is frequently mutated and/or amplified to drive cancerous cell growth. A cell surface receptor, it’s a target of both monoclonal antibodies and small molecule tyrosine kinase inhibitors for treatment of lung and other cancers.
The known variation in the level of upmodulation of EGFR is clearly visible in AI-generated data. The scale of the brain distribution makes the lung distribution look pretty small, but it’s there!
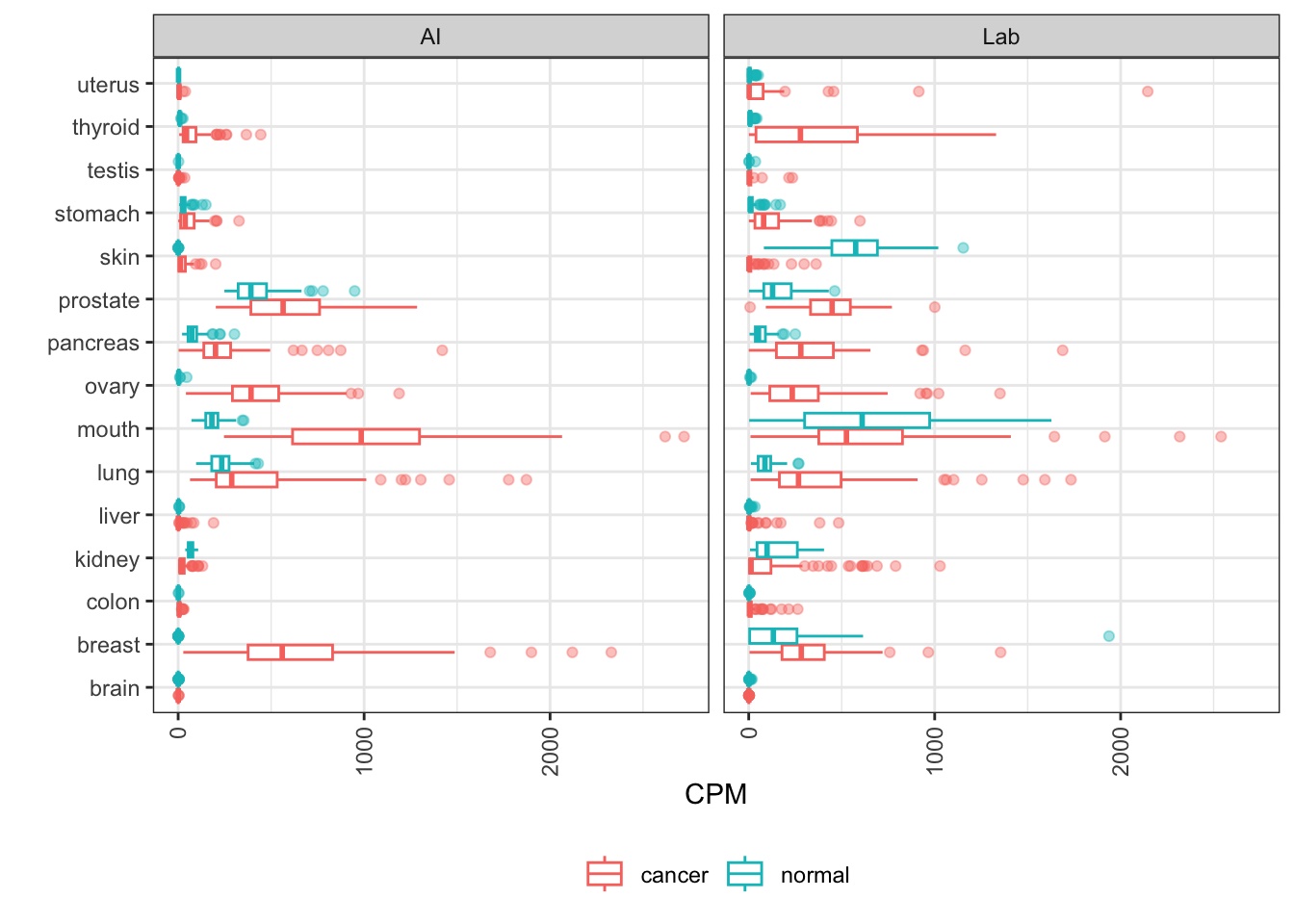
TROP2
TACSTD2, or TROP2, is an oncogene expressed in a variety of solid tumors, and a marker of poor prognosis and metastasis. Its protein, Trop-2, is the target of ADC sacituzumab govitecan which is approved for certain breast cancers.
Here the patterns are roughly recapitulated, like upmodulation in lung and thyroid. But there are some differences, like the downmodulation in skin that AI data misses. AI data confirms other indications that might be good candidates for indication expansion.
What’s next?
At Synthesize Bio, we are building the generative genomics lab for all scientists. SYNTH-cancer is one demonstration of how we envision foundation models enhancing workflows through:
- Accelerating Discovery: Our models can generate biologically relevant data for major human diseases, allowing significant expansion of available expression datasets for research.
- Augmenting Experiments: Scientists can use our platform to expand existing datasets, increasing the depth at which the more intractable diseases can be explored.
- Modeling Human Disease: The model successfully recapitulated major themes of cancer like subtying and gene set trends, demonstrating its readiness to extend the reach of science into cancer biology.
About Synthesize Bio
Synthesize Bio is building the generative genomics lab for all scientists. We believe that analyzing gene expression data without the right context is like trying to solve a puzzle with half the pieces missing. That’s why we’re developing large-scale foundation models for gene expression that take in experimental designs and output gene expression data—essentially simulating the biological experiment itself. This lets scientists prototype hypotheses faster, iterate more efficiently, and ask better questions earlier in the discovery process. But we’re not replacing the lab—we’re integrating it. Our platform seamlessly combines AI-generated expression data with real-world, lab-generated measurements, enabling a new kind of hybrid analysis that gives researchers the best of both worlds: the scale and speed of generative AI with the grounding and nuance of experimental biology.
We are in the process of releasing our Featured AI Datasets - data generated to highlight the capacity and potential of these models. We're releasing them as open source data for the community to experiment with and to explore for how they could use AI-generated data in their own processes. We previously released a GTEx-inspired AI generated population gene expression resource called SYNTH-TEx, and an interferon perturbation and lupus dataset called SYNTH-interferon. The series will continue with more Featured datasets that highlight features of our models.
A Note On Appropriate Use
The NIH has issued guidelines on the use of AI technologies in Research. While Synthesize Bio is not supported by the NIH, we do agree with these guidelines and we adhere to them. We encourage our collaborators to familiarize themselves with this too.