SYNTH-interferon: Modeling the Interferon Response from Cell Lines to Human Disease
- Can AI learn core biological processes?
- Why interferons?
- Predicting perturbations in new contexts
- Interferon Leaves a Signature — Even in AI Predictions
- Models can recapitulate known biology of human disease
- Samples cluster primarily by disease
- Global differential expression analysis includes ISGs
- Gene Set Enrichment Analysis confirms type 1 interferon as a top perturbation signature
- What’s next?
- Materials and Methods
- About Synthesize Bio
SYNTH-interferon: Modeling the Interferon Response from Cell Lines to Human Disease

Can AI learn core biological processes?
In this post, we share SYNTH-interferon, a dataset that explores how models can learn about core biological pathways and be applied from simpler to more complex research questions.
We had three main goals:
- To test if our AI model could mimic lab data it already knew. We found that it could reproduce gene expression patterns for interferon alpha-treated samples from its training data.
- To test if our AI model could correctly predict how new cell lines would react to interferon alpha treatment. When we challenged the model with completely new data, it always predicted the correct direction of response, although it sometimes underestimated the strength of the reaction.
- To test if the model could capture the complexity of a real disease. We found that AI-generated data from Systemic Lupus Erythematosus (SLE) patients showed telltale signs of interferon stimulation, just like experimental data from actual patients.
Our results underscore that computationally generated data can recapitulate biologically and clinically relevant transcriptional programs and highlights the potential of AI modeling for bridging gaps where experimental data may be limited or context-specific.
Why interferons?
Type I interferons are a small group of highly related cytokines that signal through a cell-surface receptor present in a broad array of cell types and upmodulate expression a large set of target genes, termed interferon-stimulated genes (ISGs), discovered over forty years ago. Although incompletely characterized, they are known to play a central role in both antiviral defense and the pathogenesis of autoimmune diseases such as systemic lupus erythematosus (SLE). A hallmark of SLE is the persistent overexpression of ISGs, commonly referred to as the type I interferon signature, which correlates with serological disease activity and severity.
Teaching the Model Interferon: A First Test
We first tested whether the model can accurately reproduce data it saw during training. Using five datasets with control and interferon alpha-treated samples, we asked the model to generate a control sample from text metadata, then generate a perturbed sample by applying the interferon alpha signal to the AI-generated control.
Each dataset was normalized independently using edgeR’s cpm() function with TMM correction and log(CPM) output. To visualize the data, we ran PCA on the 5,000 most variable genes from the lab data - revealing, as expected, that study differences dominate.


To assess whether the model is able to capture interferon-specific transcriptomic changes, we cluster control and interferon alpha-treated samples from both lab- and AI-generated studies on the expression of genes in the Reactome pathway “Interferon alpha/beta signaling.” Our heatmap clusters reveal that for the most part, control samples and interferon alpha-treated samples cluster together regardless of lab- or AI-generated status. We see one cluster of (AI and lab) control samples with higher expression of interferon alpha signaling genes than most of the other control samples, which could be because those samples represent a derived B cell line.

We next take a closer look at how our AI-generated control and perturbed samples were able to emulate the distribution of expression of a handful of genes known to be upregulated in interferon alpha signaling. The bottom row of the ridge plot shows the expression distribution of control and interferon alpha-treated samples in each lab study, while the top row shows the expression distribution of the AI samples generated with matching metadata. Comparing these rows shows us that the model has learned to upregulate these core interferon signalling genes, and the general shape of their distribution in response to perturbation with interferon alpha treatment within each context.
Analyzing the same set of core interferon genes across all studies, we observe that the model generally approximates the average expression accurately for both control and interferon alpha-treated samples.

Predicting perturbations in new contexts
Next, we asked whether the model could predict the effects of a perturbation in entirely new contexts. For example, cases where a scientist has only profiled an untreated (control) sample.
Our models are able to generate perturbed samples from a reference sample (you provide counts from the reference sample and perturbation metadata).
We refer to two scenarios throughout this notebook:
- Lab→Lab: both control and treated samples are experimentally measured
- Lab→AI: control sample is experimentally measured, while perturbation sample is AI-generated
Using two datasets that the model did not see in training, we gave the model experimentally-generated control counts and asked it to predict expression after interferon alpha treatment (Lab→AI).
A PCA of the results shows the first principal component separates studies. PRJNA1017856 (derived myobundles, purple) is distinct from PRJNA1138251 (multiple cell lines, yellow/orange/red). Within each cell line group, model-predicted perturbed samples (from lab controls) cluster separately from lab-measured control samples.

For our core interferon signalling genes, we plot the expression distribution of control and interferon alpha-treated samples In the heldout context, we tend to underestimate the effect interferon alpha has on the expression of these genes, but the effect is consistently in the correct direction.

We check the underestimation of this expression in PRJNA1138251 in a boxplot broken out by cell line. We observe that our model has predicted core interferon gene expression more accurately for Calu-3 interferon alpha-treated cells than for A-549, which is slightly more accurate than Huh-7.5.

Interferon Leaves a Signature — Even in AI Predictions
We compare differential expression and enrichment results between AI-generated and experimental data using held-out data from A-549, Calu-3, and Huh-7.5 cell lines. For HC-1 and HEK293 cells, we only have control data; here, we apply the model to generate the treated condition (Lab→AI).
Differential expression was calculated with DESeq2 and enrichment was calculated for transcription factor motifs (TF) and Reactome pathways (REAC) with a Fisher's one-tailed test.
The Lab→AI and Lab→Lab results are not identical for A-549, Calu-3, Huh-7.5, but in each case, the Lab→AI studies consistently finds interferon signalling pathways to be enriched in upregulated genes, and share several enriched transcription factor motifs with the lab study. In the case of HC-1 and HEK293 cells, we do not have lab data treated with interferon alpha; however, we still find enriched interferon pathways and transcription factor motifs shared with other cell line responses.




Across all Lab→Lab and Lab→AI studies in this section, 20 genes were found in the top 200 significantly differentially expressed genes by log fold change. Almost all are interferon-stimulated genes.

Models can recapitulate known biology of human disease
Motivated by our positive results from cell lines, we then tested our model’s ability to model the interferon response in human immune disease. Systemic lupus erythematosus (SLE) is an autoimmune disease marked by upregulation of type 1 interferon-stimulated genes (ISGs), so it serves as an ideal test case. We compared AI-generated SLE or healthy blood gene expression data (v2 sample generation mode of blood samples with normal random ages 35±15 yrs) with two similar published RNA-seq studies from primary patient materials.
Sample sizes of healthy and SLE subjects across studies:

Samples cluster primarily by disease
A key criterion of AI-generated data quality is that it should look foremost like a lab sample, with features of scientific interest dominating over any fingerprints of the source of the data, AI or laboratory. We can explore this issue by clustering of SLE and healthy samples from each of these sources together, and we see that they cluster primarily by disease status.

Global differential expression analysis includes ISGs
Differential expression analysis of SLE vs. healthy shows that ISGs, highlighted in red, are expressed significantly higher in both AI-generated and lab-generated SLE blood samples.
Interferon-stimulated genes (ISG) are upmodulated in all three datasets.

Selected ISGs show comparable upmodulation across the studies:

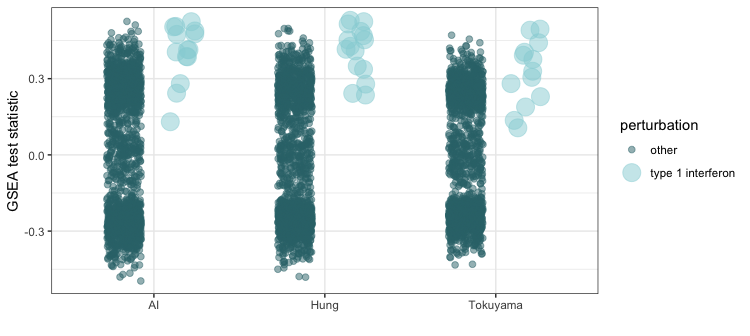
Gene Set Enrichment Analysis confirms type 1 interferon as a top perturbation signature
These data suggest that interferon induced genes are significantly upmodulated both in the AI data and in lab-generated data. To objectively test this hypothesis, we compare the expression contrast data generated for these studies with Connectivity Map’s (CMAP) database of cytokine-induced gene expression changes. We find a strong enrichment in perturbations from type 1 interferons across all three studies.
What’s next?
At Synthesize Bio, we are building the generative genomics lab for all scientists. SYNTH-interferon is one demonstration of how we envision foundation models enhancing workflows through:
- Accelerating Discovery: Our models can generate biologically relevant data for cellular perturbations, allowing researchers to explore more conditions computationally.
- Augmenting Experiments: Scientists can use our platform to predict the outcome of a treatment on their own control samples, helping to prioritize and design more efficient lab experiments.
- Modeling Human Disease: The model successfully recapitulated the interferon signature in SLE, paving the way for in silico studies of complex diseases and potentially identifying novel therapeutic targets.
Materials and Methods
Expression Data
Derived B-cell line perturbation study:
DRP011641 (bioproject: PRJDB15952) Ueda MT, Inamo J, Miya F, Shimada M, et al. Functional and dynamic profiling of transcript isoforms reveals essential roles of alternative splicing in interferon response. Cell Genom. 2024 Oct 9;4(10):100654. doi: 10.1016/j.xgen.2024.100654. Epub 2024 Sep 16. PMID: 39288763
Huh HCC cell line perturbation study:
ERP129041 (bioproject: PRJEB44928) Cheroni C, Manganaro L, Donnici L, Bevilacqua V, et al. Novel interferon-sensitive genes unveiled by correlation-driven gene selection and systems biology. Sci Rep. 2021 Sep 10;11(1):18043. doi: 10.1038/s41598-021-97258-8. Erratum in: Sci Rep. 2021 Sep 30;11(1):19870. doi: 10.1038/s41598-021-99452-0. PMID: 34508139
Pancreas perturbation studies:
SRP211834 (bioproject: PRJNA550411) and SRP255175 (bioproject: PRJNA622979) Colli ML, Ramos-Rodríguez M, Nakayasu ES, Alvelos MI, et al. An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat Commun. 2020 May 22;11(1):2584. doi: 10.1038/s41467-020-16327-0. PMID: 32444635
Lupus skin study:
SRP451611 (bioproject: PRJNA996167) Shoffner-Beck SK, Abernathy-Close L, Lazar S, Ma F, et al. Lupus dermal fibroblasts are proinflammatory and exhibit a profibrotic phenotype in scarring skin disease. JCI Insight. 2024 Feb 15;9(6):e173437. doi: 10.1172/jci.insight.173437. PMID: 38358820
Derived myobundle perturbation study:
SRP460771 (bioproject: PRJNA1017856)
A-549/Calu-3/Huh-7.5 cell line perturbation study:
SRP521433 (bioproject: PRJNA1138251)
Studies for HC-1, HEK293 cell controls:
SRP099077 (bioproject: PRJNA371822) SRP174508 (bioproject: PRJNA511908) SRP155560 (bioproject: PRJNA483230) SRP136364 (bioproject: PRJNA445481) SRP456751 (bioproject: PRJNA1008690) SRP464813 (bioproject: PRJNA1024427) SRP368405 (bioproject: PRJNA824995) SRP241725 (bioproject: PRJNA600896) SRP135771 (bioproject: PRJNA438473) SRP193326 (bioproject: PRJNA534074) SRP327795 (bioproject: PRJNA745334) SRP261001 (bioproject: PRJNA631548) SRP156596 (bioproject: PRJNA484982) SRP335073 (bioproject: PRJNA759272) SRP444879 (bioproject: PRJNA985774) SRP399908 (bioproject: PRJNA884760) SRP080966 (bioproject: PRJNA336449)
Lupus blood studies:
SRP168421 (bioproject: PRJNA505280) Tokuyama M, Kong Y, Song E, Jayewickreme T et al. ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc Natl Acad Sci U S A 2018 Dec 11;115(50):12565-12572. PMID: 30455304
SRP062966 (bioproject: PRJNA294187) Hung T, Pratt GA, Sundararaman B, Townsend MJ et al. The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science 2015 Oct 23;350(6259):455-9. PMID: 26382853
About Synthesize Bio
Synthesize Bio is building the generative genomics lab for all scientists. We believe that analyzing gene expression data without the right context is like trying to solve a puzzle with half the pieces missing. That’s why we’re developing large-scale foundation models for gene expression that take in experimental designs and output gene expression data—essentially simulating the biological experiment itself. This lets scientists prototype hypotheses faster, iterate more efficiently, and ask better questions earlier in the discovery process. But we’re not replacing the lab—we’re integrating it. Our platform seamlessly combines AI-generated expression data with real-world, lab-generated measurements, enabling a new kind of hybrid analysis that gives researchers the best of both worlds: the scale and speed of generative AI with the grounding and nuance of experimental biology.
We are in the process of releasing our Featured AI Datasets - data generated to highlight the capacity and potential of these models. We are releasing them as open source for the community to experiment with and to generate ideas for how they could use AI generated data in their own processes. We previously released an AI generated population gene expression resource called SYNTH-TEx based on the experimental design of the GTEx study. As part of our series we will be releasing other Featured datasets that highlight features of our models.