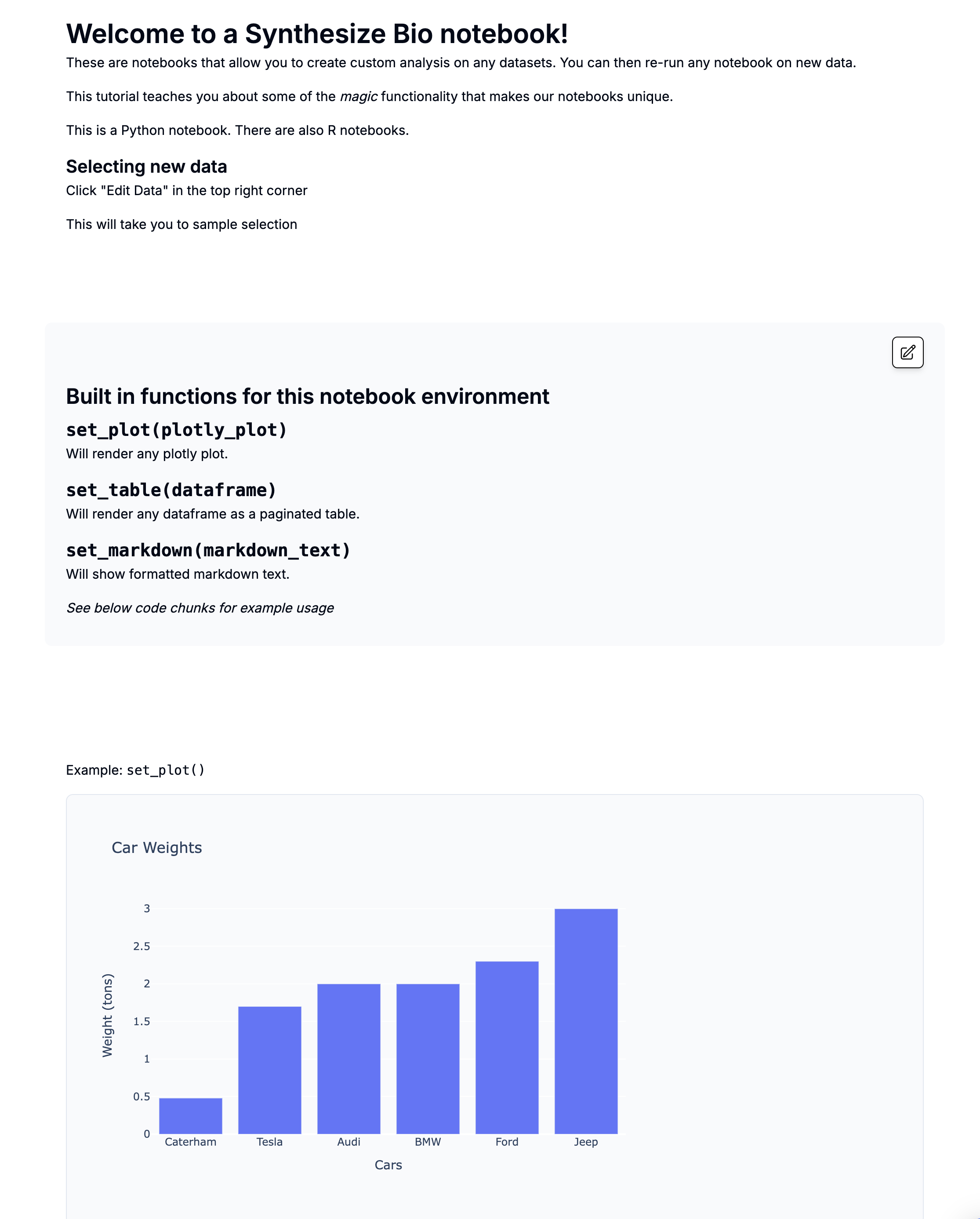
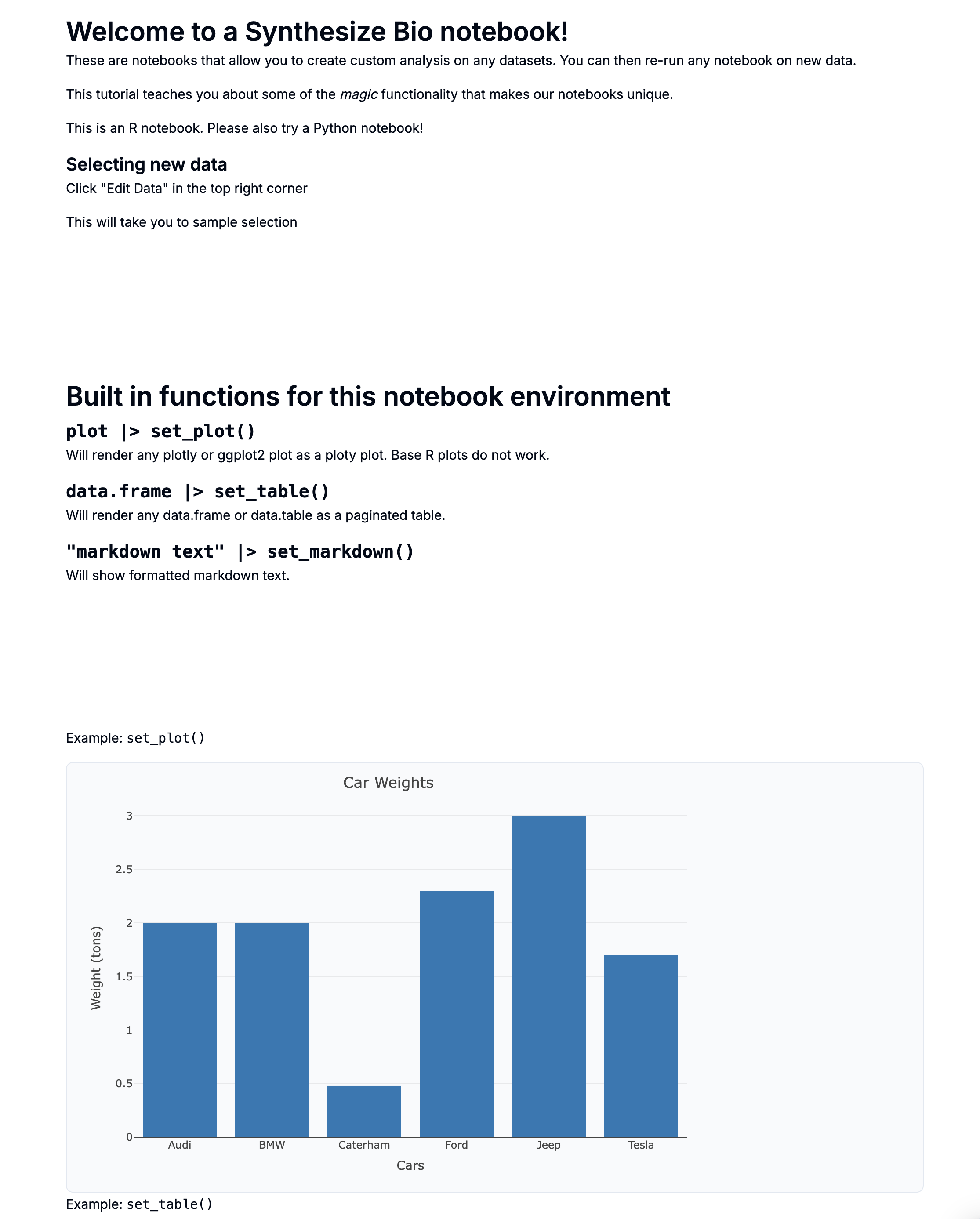
Single Cell Differential Expression with Seurat
- Seurat tutorial
- Assign Metadata
- Quality Controls
- Normalization
- Dimension Reduction
- UMAP (Uniform Manifold Approximation and Projection)
- Pairwise differential expression (two groups)
- Volcano plot of DE results
- MA plot (mean expression vs log2FC)
- Heatmap of top markers
- Dot plot for marker panel
- Violin plots for standout genes
[TEMPLATE] Single Cell Differential Expression with Seurat
This is a preview with example data.
Seurat tutorial
To work with our data we will use the Seurat package (Satija and Farrell et al. 2015).
In this tutorial we will work through similar steps as performed in this Seurat tutorial.
We will also perform a differential expression analysis to identify genes that differ between groups defined by group_label.
Now we need to import the data we selected using the load_data() function.
We'll follow up by converting this to a Seurat object by inputting the
counts into CreateSeuratObject. Lastly, we can print what we have.
Assign Metadata
We should ensure that metadata is properly set on the Seurat object.
Quality Controls
Single cell data should be QC'ed to ensure we're working with data that's sufficiently high quality.
- Unique gene count: Cells with very few genes may be low-quality or empty droplets, while those with an unusually high count could be cell doublets.
- Total molecule count: This is closely related to the unique gene count and helps identify low-quality cells.
- Percentage of mitochondrial reads: A high percentage of reads mapping to
the mitochondrial genome often indicates low-quality or dying cells. The
PercentageFeatureSet()function is used to calculate this metric by looking at all genes starting with "MT-".
Normalization
Single‑cell expression data contains noise and technical variability (e.g., library size and capture efficiency). As with any expression dataset, it should be normalized before downstream analyses.
Dimension Reduction
Single cell is high-dimensional data both in genes and cells, so dimension reduction techniques can be a useful way to explore it.
To do principal components analysis (PCA), we first ensure the data is comparable by scaling the mean log expression of each cell to 0 and the standard deviation to 1.
UMAP (Uniform Manifold Approximation and Projection)
UMAP is a non‑linear dimensionality reduction method that preserves local neighborhoods to visualize cells in 2D/3D. The Seurat package has built‑in UMAP via FindNeighbors and RunUMAP (on top PCs), with DimPlot for visualization.
Pairwise differential expression (two groups)
Run FindMarkers (Wilcoxon by default) to compare the two group_label
identities. Results report effect sizes (log2 fold change) and significance
(adjusted p-values).
Volcano plot of DE results
Visualize log2 fold change versus −log10 adjusted p-value to highlight significantly up- and down-regulated genes between the two groups, optionally labeling top hits.
MA plot (mean expression vs log2FC)
Plot average expression against log2 fold change to assess effect sizes across expression levels and check for expression-dependent biases.
Heatmap of top markers
Show scaled expression of the top up- and down-regulated genes per group
using ScaleData and DoHeatmap, grouped by group_label to reveal clear
expression patterns.
Dot plot for marker panel
Summarize percent of cells expressing and average expression per group with
DotPlot for a concise panel of selected marker genes.
Violin plots for standout genes
Display per-cell expression distributions across groups using
VlnPlot to examine heterogeneity and separation for top differentially
expressed genes.